|
Местный
Регистрация: 12.03.2018
Сообщений: 246
Спасибо: 0
Спасибо 6 в 5 постах
Репутация: 10
|
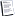
Продолжим.
Фосфатидилинозитол-3-киназа (PI3K) представляет собой гетеродимерный белок с регуляторной субъединицей 85 кДа и каталитической субъединицей 110 кДа ( PIK3CA ). PI3K служит для фосфорилирования ряда мембранных фосфолипидов. Чаще всего PI3K активируется посредством связывания лиганда с его родственным рецептором, в результате чего р85 связывается с фосфорилированными остатками тирозина на рецепторе через домен Src-homology 2 ( SH2). После ассоциации с рецептором каталитическая субъединица р110 затем переносит фосфатные группы на вышеупомянутые мембранные фосфолипиды. Именно эти липиды, особенно PtdIns(3,4,5)P3 , привлекают ряд киназ к плазматической мембране, тем самым инициируя сигнальный каскад.
Ниже PI3K находится первичная эффекторная молекула сигнального каскада PI3K, Akt/ протеинкиназа B (PKB); Akt активируется посредством фосфорилирования двух остатков: T308 и S473.
Фосфотидилинозитид-зависимые киназы ( PDK ) ответственны за активацию Akt . PDK1 является киназой, ответственной за фосфорилирование T308. Akt также фосфорилируется mLST8 (возможным PDK2), нечувствительным к рапамицину компаньоном комплекса mTOR / mTORC2. Следовательно, фосфорилирование Akt несколько затруднено, так как он фосфорилируется комплексом, расположенным ниже самого активированного Akt.
После активации Akt покидает клеточную мембрану для фосфорилирования внутриклеточных субстратов, а также способен перемещаться в ядро, где он влияет на активность ряда регуляторов транскрипции, например, CREB, E2F, NF-кВ (через IКК), транскрипционные факторы вилки и MDM2, который регулирует активность p53.
Главный ингибитор пути PI3K PTEN. Ген PTEN кодирует липидную и протеиновую фосфатазу, первичным липидным субстратом которой является PtdIns( 3,4,5 ) P3. Впрочем субстраты PTEN более разнообразны, включая киназу фокальной адгезии (FAK), обменный белок Shc и регуляторы транскрипции ETS-2 и Sp1, а также PDGFR.
Другим негативным регулятором PI3K-пути являются фосфатазы PHLPP (PH domain and Leucine rich repeat Protein Phosphatases). PHLPP (PHLPP1 и PHLPP2) дефосфорилирует Ser-473 (гидрофобный мотив) в Akt, тем самым частично инактивируя киназу. Кроме того PHLPP может дефосфорилировать киназы AGC, такие как S6K и PKC на их гидрофобных мотивах, а также ингибирующий сайт проапоптотической киназы Mst1, что приводит к ее активации.
PHLPP способен подавлять ацетилирование и фосфорилирование гистонов, чтобы уменьшить экспрессию генов рецепторных тирозинкиназ, таких как EGFR. Кроме того, PHLPP1 дефосфорилирует RAF1, обеспечивая еще один путь подавления передачи сигналов MAPK .
Две другие фосфатазы, SHIP-1 (инозитол - 5'- фосфатаза, содержащая домен SH1) , и SHIP-2 удаляют 5-фосфат из фосфолипида PtdIns(3,4,5) P 3 с образованием PtdIns(3,4) P 2. Мутации этих фосфатаз, устраняющие их активность, также могут приводить к опухолевой прогрессии.
Как известно, главной функцией Akt является ингибирование комплекса TSC1/TSC2, блокирующего белок Rheb, связывающий/обменяющий GTP, который играет ключевую роль в регуляции mTORC1 и контроле эффективности трансляции белка.
В присутствии питательных веществ mTORC1 рекрутируется на поверхность лизосом с помощью RAPTOR и активированных Rag GTPases. Оказавшись на лизосомальной мембране, mTORC1 параллельно активируется Rag GTPases вместе с Rheb GTPase. Сама Rheb GTPase представляет собой точку регуляции, поскольку ее активность регулируется комплексом TSC1/TSC2, который управляет гидролизом GTP до GDP в Rheb, тем самым вызывая его инактивацию. Активность TSC1/TSC2 регулируется множеством сигналов окружающей среды, включая присутствие фактора роста и уровня энергии клетки.
Кстати Rheb является мишенью FTI (ингибиторов фарнезилтрансферазы), с помощью которой изопренильная группа добавляется к остаток цистеина. Это важный процесс, обеспечивающий белок-белковые и белок-мембранные взаимодействия. Эти ингибиторы были разработаны с целью нацеливания на Ras. К сожалению, клинические испытания ингибиторов FT (FTI) дали неутешительные результаты. Отсутствие полезности FTI может быть связано с несколькими причинами. Во-первых, есть много белков, которые регулируются FT. Во-вторых, хотя FT модифицирует чаще всего H-Ras и в меньшей степени K-Ras, N-Ras также может модифицироваться геранил - геранил - трансферазой (ГГТ). Этот модифицированный N-Ras по-прежнему способен поддерживать биологическую потребность в Ras в раковой клетке.
Но некоторый клинический эффект FTI был связан с блокадой Rheb. Дело в том, что Rheb располагается в различных эндомембранных структурах, включая ER и Golgi, причем обнаруживается в основном в цитозольной фракции, что свидетельствует о слабом взаимодействии с мембранами.
Фарнезилирование Rheb делает возможным его слабое взаимодействие с мембраной ER и что это временное и обратимое взаимодействие необходимо для активации mTORC1. В случае Rheb на мембранах Гольджи предполагается, что непосредственная близость с mTORC1 на лизосомальных мембранах опосредуется через взаимосвязанные поверхности между этими органеллами.
mTORC1 представляет собой S/T-киназу с молекулярной массой 289 кДа. Он регулирует трансляцию в ответ на питательные вещества и факторы роста путем фосфорилирования компонентов механизма синтеза белка, включая p70 S6K и эукариотический фактор инициации трансляции 4EBP-1 , последний приводит к высвобождению фактора эукариотической инициации-4E (eIF-4E), позволяя eIF-4E участвовать в сборке комплекса инициации трансляции.
mTOR контролирует несколько стадий, участвующих в синтезе белка, но, что также важно, усиливает продукцию ключевых молекул, таких как c-Myc, cyclin D1, p27 Kip1 и белок ретинобластомы (pRb). mTOR также контролирует трансляцию мРНК HIF-1α, активация которого приводит к увеличению экспрессии ангиогенных факторов, таких как VEGF и PDGF. Более того, HIF-1α регулирует гликолитический путь, контролируя экспрессию молекул, чувствительных к глюкозе, включая переносчики глюкозы Glut 1 и Glut3.
Многие из мРНК, кодирующих ранее упомянутые гены, содержат 5'- нетранслируемые области , которые богаты G+C и трудно транслируются и поэтому называются слабыми мРНК. 4EPB-1 образует комплекс с этими мРНК и др. связывающими факторами, что делает возможным трансляцию этих слабых мРНК. ERK, p90 Rsk-1 , MNK1/2 и p70S6K регулируют фосфорилирование многих белков, участвующих в ключевом комплексе, необходимом для трансляции слабых мРНК. Mcl-1 является примером слабой мРНК и играет ключевую роль в регуляции апоптоза. Ингибиторы рапамицина и киназы mTOR подавляют трансляцию этих критических мРНК, участвующих в выживании и росте клеток.
Недавние данные показали, что в солидных опухолях ингибирование mTORC1 приводит к активации ERK 1/2 посредством p70 S6K /PI3K/Ras/Raf/MEK. Также mTORC2 может функционировать как неуловимая PDK-2, которая фосфорилирует Akt на S473 в ответ на стимуляцию фактором роста. Предполагается, что между этими двумя комплексами существует равновесие; когда образуется комплекс mTORC1, это может препятствовать образованию комплекса mTORC2 и снижать активность Akt.
|